Локализация позиций примесей углерода в кристаллических структурах полиморфных модификаций Nb5Si3 по данным атомистического компьютерного моделирования
Методом атомистического компьютерного моделирования разработана согласованная модель потенциалов межатомного взаимодействия, позволившая воспроизвести кристаллические структуры трех полиморфных модификаций силицида ниобия (α-Nb5Si3, β-Nb5Si3, γ-Nb5Si3) с ошибкой не более 0,6%. Для оценки энергетики вхождения примесных атомов углерода в структуры дополнительно разработаны парные потенциалы межатомного взаимодействия Si–C, Nb–C и C–C. Двумя независимыми методами (Мотта–Литтлтона и методом моделирования в сверхъячейках) впервые определены энергетически наиболее выгодные позиции для примесных атомов углерода в исследуемых структурах.
Введение
В настоящее время наблюдается высокая эффективность применения компьютерных технологий во всех областях науки и техники. Применение методов вычислительной математики не только позволяет избавить исследователя от рутинного «ручного» труда, но и найти факты и закономерности, не доступные современному эксперименту. Знание оптимального набора межатомных потенциалов позволяет корректно описать такие характеристики кристаллов, как энергия сцепления, механические, термодинамические и многие другие свойства. Ряд этих свойств не всегда может быть измерен экспериментально. В связи с этим атомистические расчеты с использованием оптимизированных значений межатомных потенциалов востребованы для решения широкого круга задач материаловедения. Важно отметить, что в атомистическом моделировании достаточно часто (и успешно) применяется принцип «трансферабельности» межатомных потенциалов. Он заключается в том, что оптимизированный на некоторой простой и изученной структуре набор параметров потенциалов используется без существенных изменений для моделирования более сложных (часто даже гипотетических) соединений. Таким образом удается получить неизвестную из эксперимента информацию об их структуре и свойствах [1].
Исследование и разработка материалов с принципиально новыми свойствами на основе методов атомно-молекулярного конструирования (совместно с ведущими университетами) формирует научные подходы к созданию новых материалов, оптимизирует экспериментальные исследования, позволяет получить результат с существенной экономией ресурсов и времени, обеспечивая прогнозирование и планирование действий в области создания материалов [2].
Одной из приоритетных задач материаловедения является создание супержаропрочного материала с низкой плотностью 7,2–7,5 г/см3, работоспособного при температуре до 1350°С. В качестве альтернативного жаропрочного материала будущего, который должен заменить монокристаллические никелевые жаропрочные сплавы при производстве лопаток перспективных ГТД с повышенными эксплуатационными параметрами и надежностью, рассматривают высокотемпературный естественный композит на основе ниобия, упрочненный силицидом ниобия [3–7].
Силициды ниобия являются армирующей фазой в естественно композиционных материалах на основе легированной системы Nb–Si. Жаропрочность таких композитов при высоких температурах в интервале 1200–1350°С определяется свойствами силицидов. Известны три полиморфных модификации Nb5Si3, кристаллизующиеся в различных структурных типах (табл. 1).
Таблица 1
Характеристики исследуемых полиморфных модификаций Nb5Si3
Модификация, пространственная группа симметрии | Параметры элементарной ячейки, нм | Структурный тип |
α, I4/mcm [8] | α=0,6570; c=1,1884 | Cr5B3 |
β, I4/mcm[9] | α=1,0018; c=0,5072 | Mo5Si3 |
γ, P63/mcm[10, 11] | α=0,7536; c=0,5249 | Mn5Si3 |
Для исследования структурных особенностей каждой полиморфной модификации построены структурные модели α-, β- и γ-модификаций силицидов Nb5Si3 [12]. В предыдущих работах авторов данной статьи [13, 14] методами компьютерного моделирования проведено объемное сканирование элементарных ячеек модификаций для оценки имеющихся в кристаллических структурах пустот (пор). Определены координаты расположения всех возможных пустот в α-, β- и γ-модификациях Nb5Si3. Проведенный геометрический кристаллохимический анализ для трех модификаций силицида ниобия Nb5Si3, основанный на данных об атомных радиусах элементов, позволил выявить наиболее вероятные позиции для внедрения примесных атомов С, B, N и O.
Для определения наиболее прочных связей атомов-примесей с Nb и Si для всех возможных позиций C, N, О и В, в работах [13, 14] проанализированы полиэдры Воронова–Дирихле (ПВД) пустот в α-, β- и γ-модификациях. Проведенный анализ возможных положений атомов-примесей, обладающих наиболее сильными связями с Nb и Si, показал, что для α-Nb5Si3 модификации все 5 рассмотренных позиций являются потенциально возможными, для β-Nb5Si3 только 4 из 8 позиций обладают сильными связями, для γ-Nb5Si3 – только 5 из 12.
На основании проведенного объемного сканирования элементарных ячеек сделан вывод о возможных путях диффузии атомов внедрения бора, углерода, азота и кислорода для каждой из модификаций Nb5Si3, оценены изоморфные емкости этих модификаций. Отмечена значительная диффузионная проницаемость структуры гексагонального силицида ниобия в направлении кристаллографической оси С по сравнению с α- и β-модификациями.
Цель данной работы – уточнение геометрической формы окружения и определение наиболее энергетически предпочтительных позиций для внедрения атомов углерода в трех структурных модификациях силицидов Nb5Si3 с использованием потенциалов межатомного взаимодействия. Для решения поставленной задачи:
– разработана модель парных потенциалов межатомного взаимодействия, способная с одним и тем же набором параметров воспроизвести структуры всех трех полиморфных модификаций Nb5Si3, а также SiC, Nb6C5 и Nb4SiC3 (согласованная модель);
– полученная согласованная модель парных потенциалов применена для энергетической оценки вхождения атомов углерода в выявленные ранее возможные позиции;
– выявлены искажения структуры (конечной геометрической формы) и впервые определены энергетически наиболее выгодные позиции для примесных атомов углерода в исследуемых структурах.
Материалы и методы
Атомистическое компьютерное моделирование осуществляли с использованием программы GULP 4.0 [15]. С учетом сильно выраженного ковалентного характера межатомного взаимодействия в изучаемых структурах в используемой модели потенциалов для описания химического связывания использовался короткодействующий потенциал вида Морзе:
V(r)=D·[exp(-2σ(r-r0)-2exp(-σ(r-r0))],
где r – расстояние между атомами, нм; D – энергия разрыва связи между атомами, эВ; σ – параметр мягкости, нм-1; r0 – длина связи между атомами, нм.
Первичные расчеты проводили при температуре 273 К и без давления. Заряды на атомах считались нулевыми, так как разность значений электроотрицательности для Nb (1,6 эВ) и Si (1,9 эВ) мала, а связи в структуре Nb5Si3 являются существенно ковалентными, поэтому можно считать заряды атомов нулевыми и использовать для расчетов атомные радиусы Слэйтера.
Моделирование примесных дефектов в структурах осуществляли двумя независимыми способами:
– с использованием процедуры расчета точечных дефектов методом Мотта–Литтлтона или модели «вложенных сфер» [16];
– методом моделирования в сверхъячейках.
Первый метод позволяет рассчитать энергии образования различных нульмерных дефектов: изолированных примесей, вакансий, интерстиций и их ассоциатов. Особенность данного метода заключается в том, что точечный дефект кристаллической структуры и область вокруг дефекта участвуют в нормальной процедуре минимизации энергии межатомного взаимодействия в результате смещений всех ионов в пределах заданной области, при этом никаких требований на электронейтральность области вокруг дефекта не накладывается. Внешняя область, где влияние смещений вокруг дефекта ничтожно, рассматривается как поляризуемый диэлектрический континуум.
В данных расчетах первая и вторая сферы действия потенциалов составили 0,85 и 1,85 нм соответственно.
Второй метод моделирования в сверхъячейках позволяет моделировать электронейтральные дефекты, дает возможность учитывать дальнее взаимодействие, но требует значительных компьютерных мощностей и времени по сравнению с методом Мотта–Литтлтона.
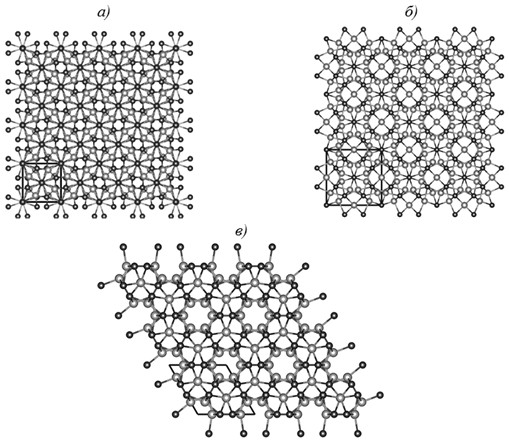
Рис. 1. Сверхъячейки 4×4×3 (768 атомов) для модификации α-Nb5Si3 (а), 3×3×5 (720 атомов) для модификаций β-Nb5Si3 (б) и γ-Nb5Si3 (в) (проекция в плоскости ab; темным цветом показаны атомы Nb, светлым – атомы Si)
Для моделирования вхождения атомов углерода в структуры Nb5Si3 сконструированы сверхъячейки: 4×4×3 (768 атомов) – для модификации α-Nb5Si3, 3×3×5 (720 атомов) – для модификаций β-Nb5Si3 и γ-Nb5Si3 (рис. 1), содержащие приблизительно равное число атомов и имеющие похожие параметры a, b и c. Визуализацию сверхъячеек кристаллических структур осуществляли с использованием компьютерной программы VESTA [17].
Результаты
Разработанная согласованная модель потенциалов межатомного взаимодействия для пар атомов Nb–Nb, Nb–Si и Si–Si (табл. 2) позволила воспроизвести параметры элементарных ячеек исследуемых модификаций Nb5Si3 с ошибкой не более 0,6% (табл. 3). Согласованность потенциалов дает возможность использовать их в дальнейшем при моделировании структур при различных значениях давления и температуры, а также при конструировании твердых растворов замещения.
Для моделирования вхождения атомов углерода в исследуемые структуры дополнительно разработаны потенциалы Морзе для пар атомов Nb–C, Si–C и C–C (табл. 2) на основе структур Nb6C5 [18], SiC [19] и Nb4SiC3 [20] с последующим уточнением значений параметров на структурах Nb5Si3.
Таблица 2
Параметры потенциалов Морзе межатомного взаимодействия,
используемые при расчетах
Пара атомов | D, эВ | σ, нм-1 | r0, нм |
Nb–Nb | 0,3501 | 0,10706 | 0,3556 |
Nb–Si | 0,535195 | 0,2575885 | 0,2688328 |
Si–Si | 0,233629 | 0,1679402 | 0,2350483 |
Nb–C | 0,040515 | 0,2238074 | 0,2330894 |
Si–C | 0,255152 | 0,1181991 | 0,2695635 |
C–C | 0,341557 | 0,1924411 | 0,1112259 |
Таблица 3
Рассчитанные параметры элементарных ячеек структур с указанием отклонения
от начальных значений для модификаций Nb5Si3
Параметр*, единица измерения | Начальное значение | Конечное значение | Разность конечного | Отклонение, % |
Модификация α-Nb5Si3 | ||||
V, нм3 | 0,51297 | 0,51346 | 0,00049 | 0,10 |
α, нм | 0,657 | 0,658 | 0,001 | 0,04 |
b, нм | 0,657 | 0,657 | 0 | 0 |
c, нм | 1,188 | 1,188 | 0 | 0 |
α, град | 90 | 90 | 0 | 0 |
β, град | 90 | 90 | 0 | 0 |
γ, град | 90 | 90 | 0 | 0 |
Модификация β-Nb5Si3 | ||||
V, нм3 | 0,509028 | 0,50969 | 0,00066 | 0,13 |
α, нм | 1,002 | 1,002 | 0 | 0 |
b, нм | 1,002 | 1,002 | 0 | 0 |
c, нм | 0,507 | 0,507 | 0 | 0 |
α, град | 90 | 90 | 0 | 0 |
β, град | 90 | 90 | 0 | 0 |
γ, град | 90 | 90 | 0 | 0 |
Модификация γ-Nb5Si3 | ||||
V, нм3 | 0,25816 | 0,25725 | -0,00091 | -0,35 |
α, нм | 0,754 | 0,754 | 0 | 0 |
b, нм | 0,754 | 0,754 | 0 | 0 |
c, нм | 0,525 | 0,522 | -0,003 | -0,58 |
α, град | 90 | 90 | 0 | 0 |
β, град | 90 | 90 | 0 | 0 |
γ, град | 120 | 120 | 0 | 0 |
* V – объем элементарной ячейки; α, b, c, α, β, γ – параметры элементарных ячеек.
В результате расчетов методом Мотта–Литтлтона вычислены энергии конфигураций областей дефектов для потенциально возможных позиций атомов внедрения углерода (табл. 4), равные разности энергий бездефектной структуры и структуры с дефектом.
Таблица 4
Характеристики параметров пустот до и после энергетической оптимизации
по методу «вложенных сфер»
Условный номер пустот | До оптимизации структуры | После оптимизации структуры | |||||
Координата атома внедрения углерода | Объем полиэдра, нм3 | Координата атома внедрения углерода | Атомная пара | Расстояния в полиэдре*, нм | Объем полиэдра, нм3 | E, эВ | |
Модификация γ-Nb5Si3 | |||||||
1 | 0; 0; 0 | 0,01614 | 0; 0; 0 | C–Nb | 0,2310 (6) | 0,01642 | 0 (0) |
2 | 0,6; 0; 0,5 | 0,00258 | 0,470; 0,154; 0,003 | C–Nb C–Si C–Si C–Nb C–Nb | 0,1934 (1) 0,2133 (1) 0,2147 (1) 0,1871 (1) 0,1860 (1) | 0,00639 | 1,14 (1,63) |
3 | 0,6; 0; 0,4 | 0,00258 | 0,468; 0,154; 0,607 | C–Nb C–Si C–Nb C–Si C–Nb | 0,1934 (1) 0,2146 (1) 0,1860 (1) 0,2132 (1) 0,1871 (1) | 0,00639 | 1,14 (1,63) |
4 | 0,6; 0,05; 0,55 | 0,00258 | 0,546; 0; 0,609 | C–Si | 0,2310 (6) | 0,01642 | 0 (0) |
5 | 0,6; 0,05; 0,75 | 0,00258 | 0,558; 0,006; 0,914 | C–Si C–Nb C–Nb C–Nb | 0,1831 (1) 0,2568 (2) 0,2562 (2) 0,1652 (1) | 0,01247 | 2,97 (3,33) |
Модификация α-Nb5Si3 | |||||||
1 | 0,35; 0,70; 0,10 | 0,00267 | 0,032; 0,054; 0,309 | C–Nb C–Si C–Nb C–Nb C–Si | 0,1810 (1) 0,2100 (1) 0,1968 (1) 0,1970 (1) 0,2260 (1) | 0,00628 | 0,74 (3,97) |
2 | 0,80; 0,70; 0,10 | 0,00284 | 0,969; 0,891; 0,467 | C–Nb C–Nb C–Nb | 0,1945 (2) 0,2315 (1) 0,1891 (1) | 0,00397 | 0 (0) |
3 | 0,85; 0; 0,40 | 0,00565 | 0,806; 0,257; 0,633 | C–Si C–Nb C–Nb C–Si C–Nb | 0,2260 (1) 0,1968 (1) 0,1971 (1) 0,2100 (1) 0,1811 (1) | 0,00628 | 0,74 (3,97) |
4 | 0,20; 0; 0,50 | 0,00267 | 0,229; 0,348; 0,942 | C–Nb C–Nb C–Si C–Nb | 0,1581 (1) 0,2414 (2) 0,1719 (1) 0,2400 (2) | 0,01225 | 3,36 (5,09) |
5 | 0,50; 0; 0,50 | 0,00852 | 0,286; 0; 0,942 | C–Nb C–Si | 0,2189 (4) 0,1683 (2) | 0,01068 | 5,39 (7,71) |
Модификация β-Nb5Si3 | |||||||
1 | 0,15; 0; 0 | 0,00523 | 0,200; 0,346; 0 | C–Si C–Nb C–Nb C–Nb | 0,170823 (1) 0,157556 (1) 0,242360 (2) 0,241720 (2) | 0,01226 | 3,18 (0,84) |
2 | 0,50; 0,10; 0,25 | 0,00573 | 0,286; 0; 0,971 | C–Nb C–Nb | 0,1988 (2) 0,1987 (2) | 0,00402 | 0 (0) |
3 | 0,65; 0,05; 0,30 | 0,00287 | 0,287; 0; 0,971 | C–Nb | 0,1988 (4) | 0,00402 | 0 (0) |
4 | 0,90; 0,40; 0,30 | 0,00298 | 0,573; 0,286; 0,971 | C–Nb | 0,1988 (4) | 0,00402 | 0 (0) |
* Параметры связей указаны между соответствующими па́рами атомов с указанием длины связи и количества связей (в скобках).
** E – энергия конфигурации области дефекта относительно наилучшего значения (в скобках указана величина, полученная методом моделирования в сверхъячейках).
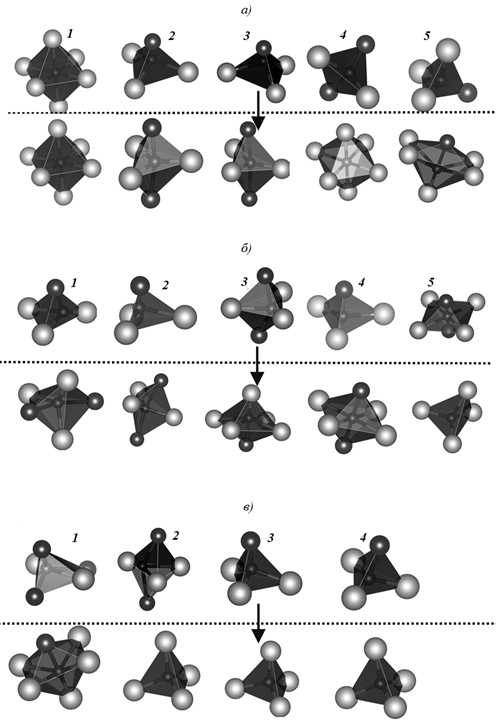
Рис. 2. Координационные полиэдры атомов внедрения углерода до энергетической оптимизации (вверху) и после (внизу) для модификаций γ-Nb5Si3 (а), α-Nb5Si3 (б) и β-Nb5Si3, (в) (светлые шары – атомы Nb, темные шары – атомы Si, 1–5 – номера пустот согласно табл. 4)
Рассмотрено изменение геометрической формы окружения атома-примеси после энергетической оптимизации. Как видно из данных табл. 4, атом углерода со стартовыми координатами (х; у; z) не меняет в результате энергетической оптимизации своей позиции и геометрической формы окружения (правильный октаэдр) в гексагональной модификации γ-Nb5Si3 также не меняются (0; 0; 0), что подтверждает предпочтительную позицию для атомов углерода в этом бесконечном канале структуры. Во всех остальных случаях конфигурация области дефекта происходит с существенным изменением начальных координат позиции примесного атома и его окружения, вплоть до смены координационного числа (рис. 2).
Обсуждение результатов
Методами атомистического моделирования впервые определены энергетически наиболее выгодные позиции для примесных атомов углерода в исследуемых структурах:
– для модификации γ-Nb5Si3 – позиция атома углерода (0; 0; 0) в октаэдрическом окружении шести атомов Nb с длиной связи Nb–C, равной 0,231 нм;
– для модификации α-Nb5Si3 – позиция атома углерода (0,979; 0,891; 0,467) в окружении атомов Nb, образующих искаженный тетраэдр с межатомными расстояниями для пары С–Nb: 0,2316 и 0,1891 нм (по одной связи); 0,1945 (две связи);
– для модификации β-Nb5Si3 – позицияатома углерода (0,0286; 0; 0,0971) в тетраэдрическом окружении четырех атомов Nb с длиной связи C–Nb, равной 0,1988 нм.
На основании вычисленных энергий дефектов, можно сделать вывод о том, что в модификации γ-Nb5Si3 имеется всего три вместо пяти геометрически возможных форм типов пустот [13], подходящих для вхождения атомов углерода в позиции с координатами (0; 0; 0), (0,470; 0,154; 0,003) и (0,558; 0,006; 0,914); для модификации α-Nb5Si3 – четыре типа пустот в позиции с координатами (0,032; 0,054; 0,309), (0,969; 0,891; 0,467), (0,229; 0,348; 0,942) и (0,286; 0; 0,942) из пяти возможных, а для модификации β-Nb5Si3 – только два типа пустот в позиции с координатами (0,200; 0,346; 0) и (0,286; 0; 0,971) из четырех геометрически возможных форм. В гексагональной структуре пустоты с координатами центра (0; 0; 0) формируют бесконечные каналы вдоль кристаллографической оси С, остальные выявленные позиции изолированы.
Расчеты величины энергий дефекта методом сверхъячеек коррелируют с расчетами методом «вложенных сфер» и с их помощью можно предсказать в качестве наилучших одни и те же позиции (табл. 4). Так, для сверхъячейки 3×3×5 модификации γ-Nb5Si3 позиции 2 и 3 хуже по энергии, чем позиции 1 и 4 – на 1,63 эВ (1,14 эВ – по методу Мотта–Литтлтона), а позиция 5 – на 3,33 эВ (2,97 эВ). Для сверхъячейки 4×4×3 модификации α-Nb5Si3 ячейки энергии дефектов 1 и 3 хуже на 3,97 эВ (0,74 эВ – по методу Мотта–Литтлтона), позиция 4 – на 5,09 эВ (3,36 эВ), а позиция 5 – на 7,71 эВ (5,39 эВ), чем энергия позиции 2 соответственно. Для сверхъячейки 3×3×5 модификации β-Nb5Si3 позиции 2, 3 и 4 на 0,84 эВ выгоднее, чем позиция 1 (3,18 эВ – по методу Мотта–Литтлтона).
Заключения
1. Разработана согласованная модель потенциалов межатомного взаимодействия, которая позволила воспроизвести структурные особенности модификаций Nb5Si3 с высокой точностью (отклонение от экспериментальных значений параметров элементарных ячеек не превышает 0,6%) и осуществить моделирование вхождения атомов углерода в исследуемые кристаллические структуры.
2. Двумя независимыми методами (методом Мотта–Литтлтона и методом моделирования в сверхъячейках: 4×4×3 (768 атомов) – для модификации α-Nb5Si3, 3×3×5 (720 атомов) – для модификаций β-Nb5Si3 и γ-Nb5Si3) оценена энергетика вхождения атомов углерода в возможные кристаллографические позиции (пустоты) модификаций Nb5Si3.
3. Полуэмпирические расчеты величин энергий конфигураций областей дефектов показали, что вхождение углерода в структуры Nb5Si3 практически во всех случаях сопровождается существенным изменением начальной геометрической формы окружения атома-примеси вплоть до смены первого координационного числа. Наиболее энергетически выгодной позицией для вхождения атома углерода в модификацию α-Nb5Si3 является искаженный тетраэдр в позиции с координатами (0,97; 0,89; 0,47), в модификацию β-Nb5Si3 – тетраэдр в позиции с координатами (0,29; 0; 0,97), в модификацию γ-Nb5Si3 – октаэдр с центром в начале координат, формирующий бесконечные каналы структуры вдоль кристаллографической оси С, обеспечивая ускоренную диффузию элемента внедрения в этом направлении.
Благодарности
Авторы признательны профессору, доктору технических наук Игорю Леонидовичу Светлову за полезную дискуссию при обсуждении диффузионных процессов в структуре силицида и результатов данного исследования.
Работа выполнена в рамках реализации комплексного научного направления 9.4. «Композиты на основе Nb–Si с повышенной стойкостью к окислению и коррозии» («Стратегические направления развития материалов и технологий их переработки на период до 2030 года») [3] и при поддержке Российского фонда фундаментальных исследований: гранты №15-05-06742 и №15-05-04575.
- Урусов В.С., Еремин Н.Н. Атомистическое компьютерное моделирование структуры и свойств неорганических кристаллов и минералов, их дефектов и твердых растворов. М.: ГЕОС, 2012. 428 с.
- Каблов Е.Н. О настоящем и будущем ВИАМ и отечественного материаловедения: интервью. URL: http://www.ras.ru/news/shownews.aspx?id=824e2453-383e-4d9e-b78d-87c9f7bf16ee (дата обращения: 31.01.2017).
- Каблов Е.Н. Инновационные разработки ФГУП «ВИАМ» ГНЦ РФ по реализации «Стратегических направлений развития материалов и технологий их переработки на период до 2030 года» // Авиационные материалы и технологии. 2015. №1 (34). С. 3–33. DOI: 10.18577/2071-9140-2015-0-1-3-33.
- Каблов Е.Н., Светлов И.Л., Ефимочкин И.Ю. Высокотемпературные Nb–Si-композиты // Вестник МГТУ им. Н.Э. Баумана. Сер.: Машиностроение. 2011. №SP2. С. 164–173.
- Тимофеева О.Б., Колодочкина В.Г., Шванова Н.Ф., Нейман А.В. Исследование микроструктуры высокотемпературного естественно композиционного материала на основе ниобия, упрочненного интерметаллидами силицида ниобия // Авиационные материалы и технологии. 2015. №1 (34). С. 60–64. DOI: 10.18577/2071-9140-2015-0-1-60-64.
- Щетанов Б.В., Ефимочкин И.Ю., Паэгле С.В., Карачевцев Ф.Н. Исследование высокотемпературной прочности in-situ-композитов на основе Nb, армированных монокристаллическими волокнами α-Al2O3 // Авиационные материалы и технологии. 2016. №3 (42). С. 53–59. DOI: 10.18577/2071-9140-2016-0-3-53-59.
- Каблов Е.Н., Мубояджян С.А. Жаростойкие и теплозащитные покрытия для лопаток турбины высокого давления перспективных ГТД // Авиационные материалы и технологии. 2012. №S. С. 60–70.
- Кочержинский Ю.А., Юпко Л.М., Шишкин Е.А. Диаграмма состояния Nb–Si // Изв. АН СССР. Металлы. 1980. №1. C. 206–211.
- Aronsson B. The crystal structure of Mo5 Si3 and W5 Si3 // Acta Chemica Scandinavica. 1955. No. 9. P. 1107–1110.
- Schachner H., Cerwenka E., Nowotny H.N. Neue Silizide vom M5Si3-Typ mit D 88-Struktur // Journal of the American Ceramic Society. 1982. Vol. 65. P. 260–265.
- Schachner H., Cerwenka E., Nowotny H. Neue Silizide vom M5Si3-Typ mit D 88-Struktur // Monatshefte fuer Chemie und verwandte Teile anderer Wissenschaften. 1954. Vol. 85. P. 245.
- Каблов Е.Н., Кузьмина Н.А., Еремин Н.Н., Светлов И.Л., Нейман А.В. Атомные модели структуры силицидов ниобия в in-situ композитах Nb–Si // Журнал структурной химии. 2017. №3. Т. 58. C. 564–570.
- Кузьмина Н.А., Еремин Н.Н., Марченко Е.И., Светлов И.Л. и др. Пути диффузии примесей внедрения в силициде ниобия Nb5Si3 различных полиморфных модификаций // Кристаллография. 2017. №4 (в печати).
- Муромцев Н.А., Кузьмина Н.А., Марченко Е.И., Еремин Н.Н., Якушев Д.А. Теоретический кристаллохимический анализ пустот в кристаллических структурах полиморфных модификаций Nb5Si3 // Минералы: строение, свойства, методы исследования: сб. тез. докл. VIII Всерос. молодежной научной конференции, Екатеринбург. 2016. С. 100–101.
- Gale G.D., Gale J.D. GULP: A computer program for the symmetry adapted simulation. Journal of the Chemical Society // Faraday Transactions. 1997. Vol. 93. P. 629–637. DOI: 10.1039/A606455H.
- Mott N.F., Littleton M.J. Conduction in polar crystals. I. Electrolytic conduction in solid salts // Transactions of the Faraday Society. 1938. Vol. 34. P. 485–495.
- Momma and Izumi F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data // Journal of Applied Crystallography. 2011. Vol. 44. P. 1272–1276.
- Khaenko B.V., Sivak O.P. Structure of the ordering of niobium monocarbide // Soviet Physics, Crystallography = Kristallografiya. 1990. Vol. 35. P. 653–655.
- Merz K.M., Adamsky R.F. Synthesis of the wurtzite form of silicon carbide. Reference // Journal of the American Chemical Society. 1959. Vol. 81 (1). P. 250–251.
- Li C.L., Kuo J.L., Wang B.A. et al. A new layer compound Nb4SiC3 predicted from first-principles theory // Journal of Physics D Applied Physics. 2009. Vol. 42. No. 7. P. 075404–075409.